国際共同第Ⅲ相試験:有効性検証試験(PATENT-1試験)
PAHに対するアデムパス®の臨床成績
国際共同第Ⅲ相試験:有効性検証試験(PATENT-1試験)
承認時評価資料:バイエル薬品社内資料[肺動脈性肺高血圧症患者を対象とした第Ⅲ相試験]
Ghofrani HA et al. N Engl J Med. 369, 330(2013)(COI:本研究はバイエル社の資金により実施された、著者にバイエル社員(3名)を含む、著者にバイエル社より講演料等を受領している者を含む)
- 試験概要
- 有効性(主要評価項目:6分間歩行距離)
- 有効性(副次的評価項目:WHO機能分類)
- 有効性(副次的評価項目:PVR)
- 有効性(副次的評価項目:NT-proBNP)
- 有効性(追加の評価項目:血行力学的パラメータ)
- 安全性
試験概要
PAH(pulmonary arterial hypertension):肺動脈性肺高血圧症
PVR(pulmonary vascular resistance):肺血管抵抗
TID:1日3回
NT-proBNP(N-terminal prohormone of brain natriuretic peptide):ヒト脳性ナトリウム利尿ペプチド前駆体N端フラグメント
†探索的用量群は比較対照群ではないため、有効性評価項目の結果は記載していない。
※ 用量調節時の指標として用いる収縮期血圧について、第Ⅲ相試験ではその基準値を“95mmHg以上”、“90~94mmHg”及び“90mmHg未満”に分けたが、[承認用法及び用量]では日常臨床においてより実用的なものにするため、“95mmHg以上”と“95mmHg未満”の2つに分けた。
6. 用法及び用量
用量調節期:通常、成人にはリオシグアトとして1回1.0mg 1日3回経口投与から開始する。2週間継続して収縮期血圧が95mmHg以上で低血圧症状を示さない場合には、2週間間隔で1回用量を0.5mgずつ増量するが、最高用量は1回2.5mg 1日3回までとする。収縮期血圧が95mmHg未満でも低血圧症状を示さない場合は、現行の用量を維持するが、低血圧症状を示す場合には、1回用量を0.5mgずつ減量する。
用量維持期:用量調節期に決定した用量を維持する。用量維持期においても、最高用量は1回2.5mg 1日3回までとし、低血圧症状を示すなど、忍容性がない場合には、1回用量を0.5mgずつ減量する。
有効性
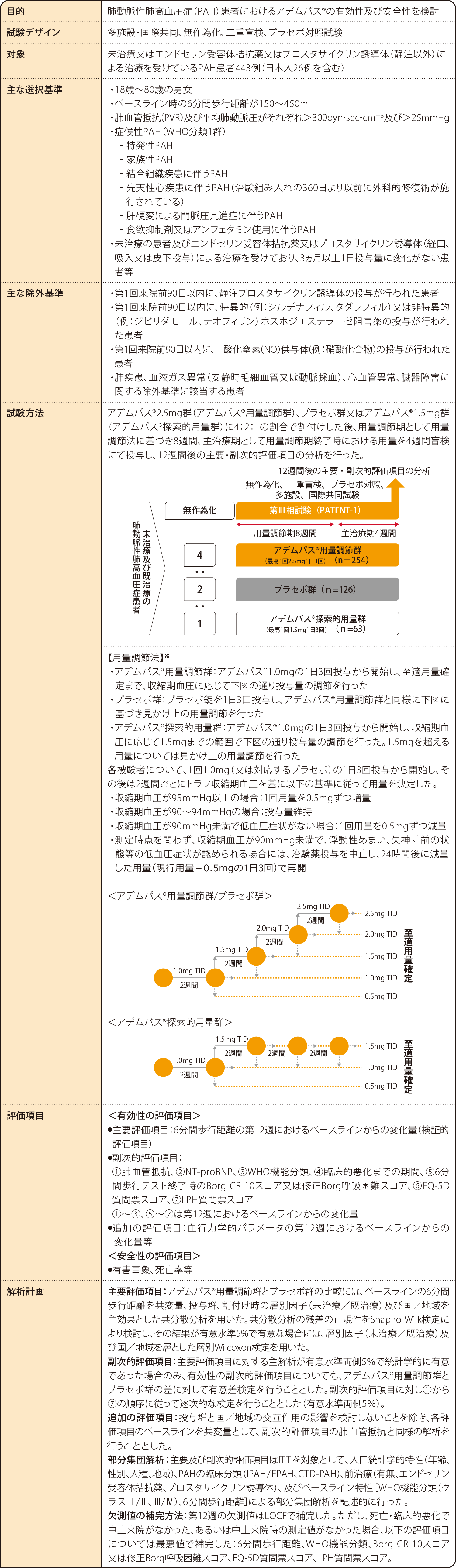
6分間歩行距離の第12週におけるベースラインからの平均変化量は、アデムパス®用量調節群29.6m、プラセボ群-5.6mであり、アデムパス®用量調節群はプラセボ群に比べて有意な改善を示しました(最小二乗平均値の差35.78[95%信頼区間:20.06~51.51]a、p<0.0001b)。
主要評価項目:6分間歩行距離のベースラインから第12週までの平均変化量(検証的解析結果)
ITT解析による評価
a: ベースライン値を共変量、投与群、割付け時の層別因子及び国/地域を主効果とした共分散分析で算出した最小二乗平均値の差及び95%信頼区間
b:層別因子(未治療/既治療)及び国/地域を層とした層別Wilcoxon検定
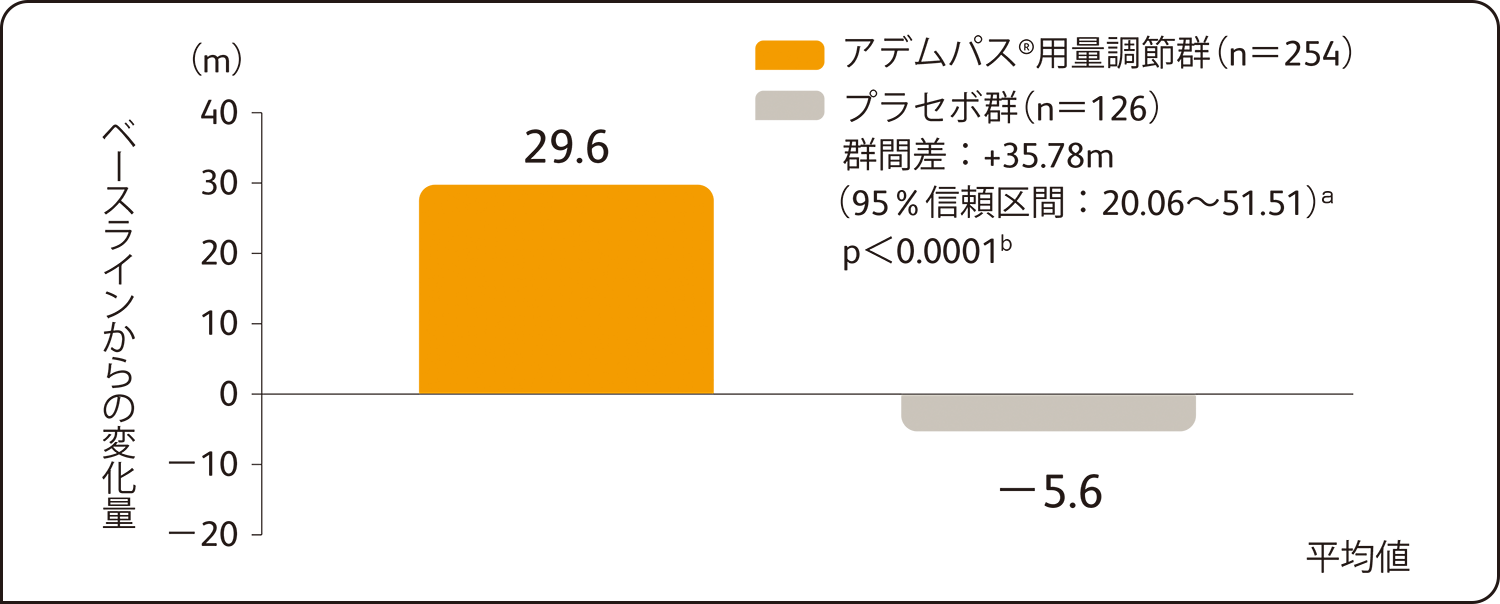
6分間歩行距離のベースラインから第12週までの変化量の推移は以下の通りでした。
6分間歩行距離のベースラインから第12週までの平均変化量の推移
ITT解析による評価
帰属値=治験を完了あるいは中止した被験者について、第12週までの最終測定値(安全性追跡来院を含まない)で補完した値。ただし、死亡・臨床的悪化で中止来院がなかった、あるいは中止来院時の測定値がなかった場合は最悪値で欠測値を補完した。
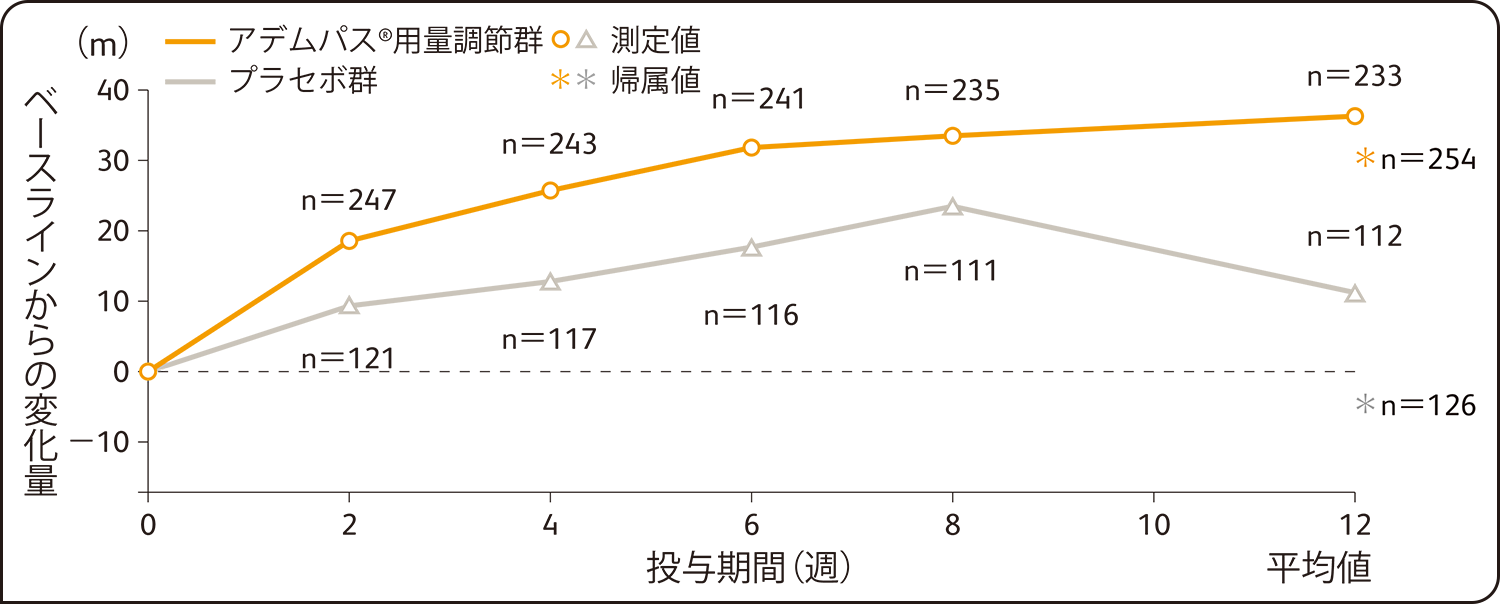
第12週においてベースラインからWHO機能分類が改善した患者の割合は、アデムパス®用量調節群で20.9%、プラセボ群で14.4%でした。第12週におけるWHO機能分類のベースラインからの変化量について、アデムパス®群はプラセボ群に比べて有意に改善しました(p=0.003a)。
副次的評価項目:WHO機能分類のベースラインから第12週までの変化量
ITT解析による評価
WHO機能分類のクラスが1段階以上改善した場合を「改善」、変化しなかった場合を「不変」、1段階以上悪化した場合を
「悪化」と分類した。投与群間の比較のための検定には、スコアの変化(治験終了時クラス-ベースライン時のクラス)を用いた。死亡をクラスⅤとし、スコアの変化は-3(ベースライン時にクラスⅣで終了時にクラスⅠ)から+4(ベースライン時にクラスⅠで終了時に死亡)までの値を取り得る。
a:層別因子(未治療/既治療)及び国/地域を層とした層別Wilcoxon検定
PVRの第12週におけるベースラインからの平均変化量は、アデムパス®用量調節群-223.29dyn・sec・cm-5、プラセボ群-8.89dyn・sec・cm-5であり、アデムパス®用量調節群はプラセボ群に比べて有意な改善を示しました(最小二乗平均値の差-225.72[95%信頼区間:-281.37~-170.08]a、p<0.0001b)。
副次的評価項目:PVRのベースラインから第12週までの平均変化量

ITT解析による評価 平均値(ベースラインからの変化量)
a:ベースライン値を共変量、投与群、割付け時の層別因子及び国/地域を主効果とした共分散分析で算出した最小二乗平均値の差及び95%信頼区間
b:層別因子(未治療/既治療)及び国/地域を層とした層別Wilcoxon検定
NT-proBNPの第12週におけるベースラインからの平均変化量は、アデムパス®用量調節群-197.89pg/mL、プラセボ群232.39pg/mLであり、アデムパス®用量調節群はプラセボ群に比べて有意な改善を示しました(最小二乗平均値の差-431.81[95%信頼区間:-781.52~-82.10]a、p<0.0001b)。
副次的評価項目:NT-proBNPのベースラインから第12週までの平均変化量
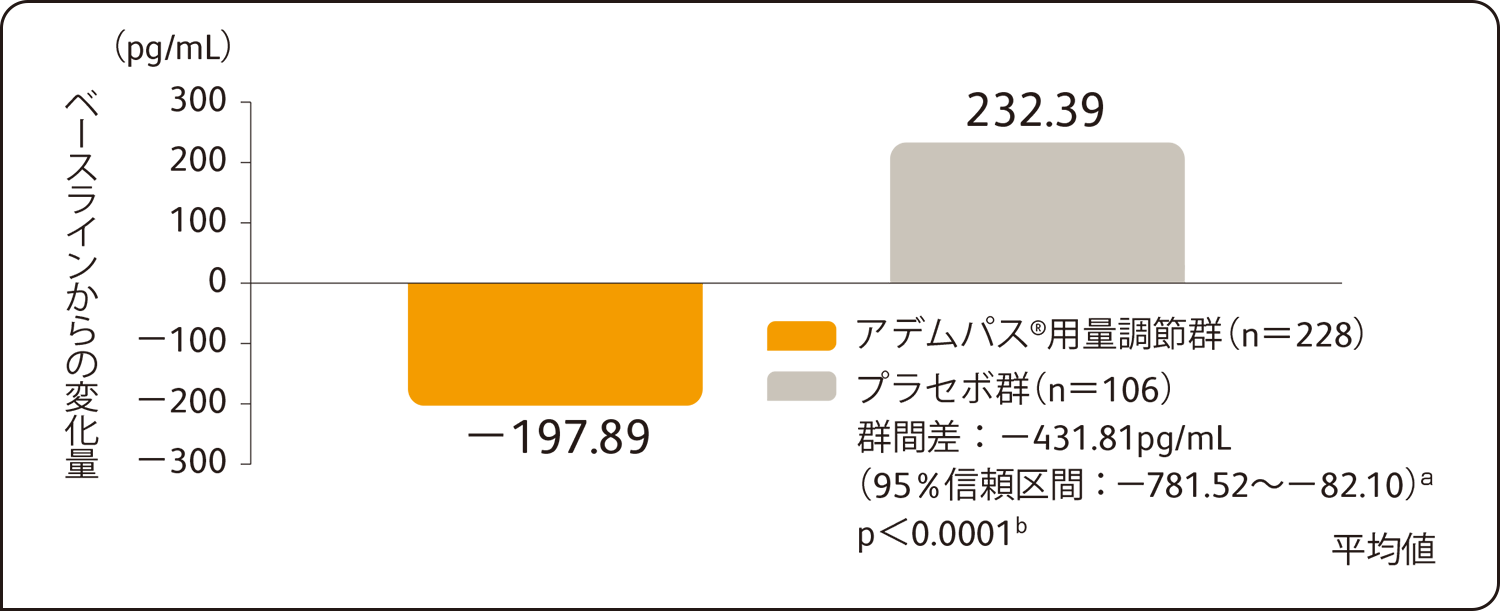
ITT解析による評価 平均値(ベースラインからの変化量)
a:ベースライン値を共変量、投与群、割付け時の層別因子及び国/地域を主効果とした共分散分析で算出した最小二乗平均値の差及び95%信頼区間
b:層別因子(未治療/既治療)及び国/地域を層とした層別Wilcoxon検定
アデムパス®用量調節群はプラセボ群に比べて、平均肺動脈圧、肺血管抵抗の有意な低下と、心拍出量及び心係数の有意な増加を示しました。
追加の評価項目:血行力学的パラメータのベースラインから第12週までの変化量におけるアデムパス®用量調節群とプラセボ群の比較
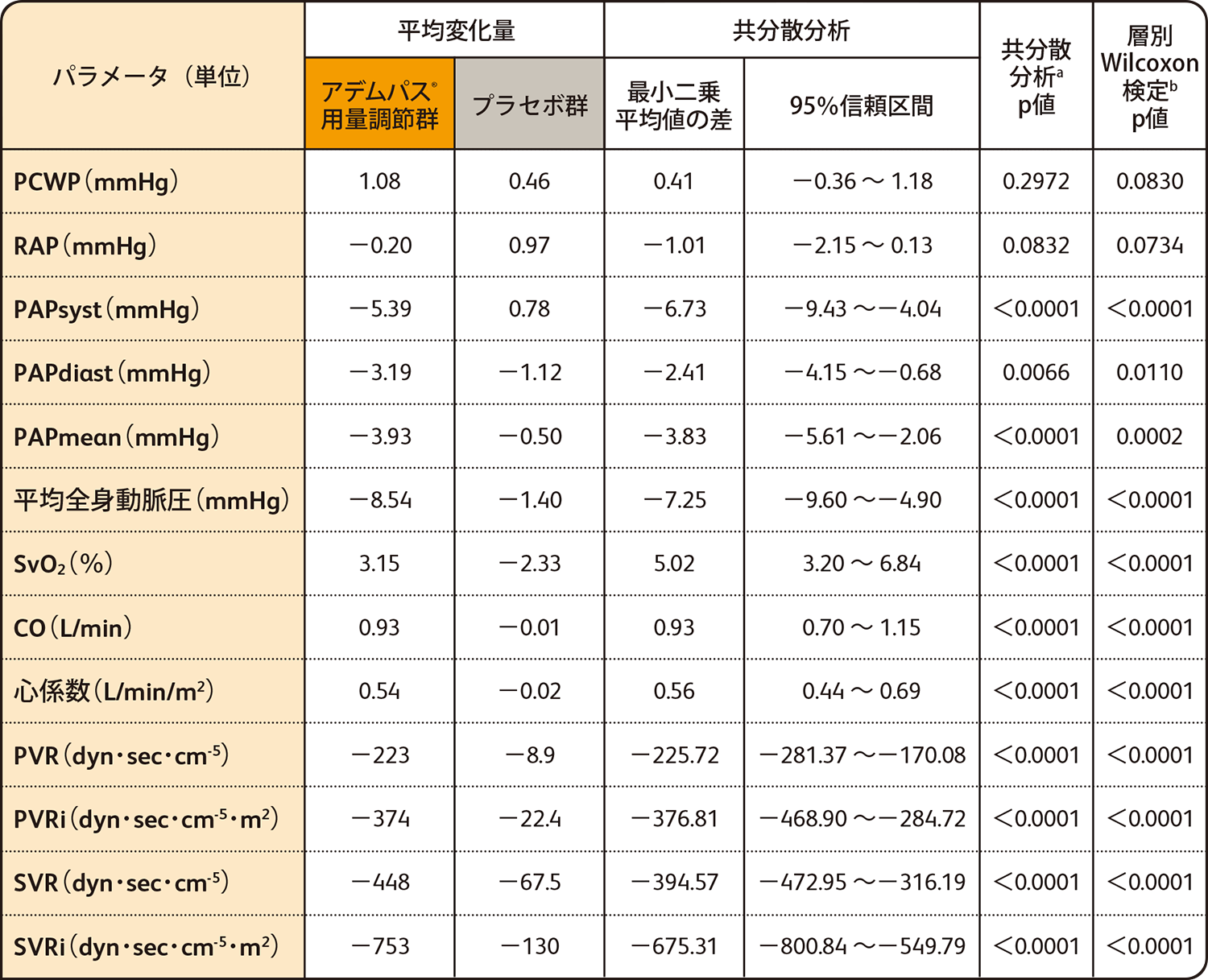
ITT解析による評価
PCWP:肺毛細血管楔入圧、RAP:右房圧、PAPsyst:収縮期肺動脈圧、PAPdiast:拡張期肺動脈圧、PAPmean:平均肺動脈圧、SvO2:混合静脈血酸素飽和度、CO:心拍出量、PVR:肺血管抵抗、PVRi:肺血管抵抗係数、SVR:全身血管抵抗、SVRi:全身血管抵抗係数
a:ベースライン値を共変量、投与群、国/地域及び割付け時の層別因子を主効果とした共分散分析
b:層別因子(未治療/既治療)及び国/地域を層とした層別Wilcoxon検定
安全性
全ての副作用発現率は、アデムパス®用量調節群で63.8%(162/254例)、プラセボ群で52.4%(66/126例)、アデムパス®探索的用量群で61.9%(39/63例)でした。
副作用
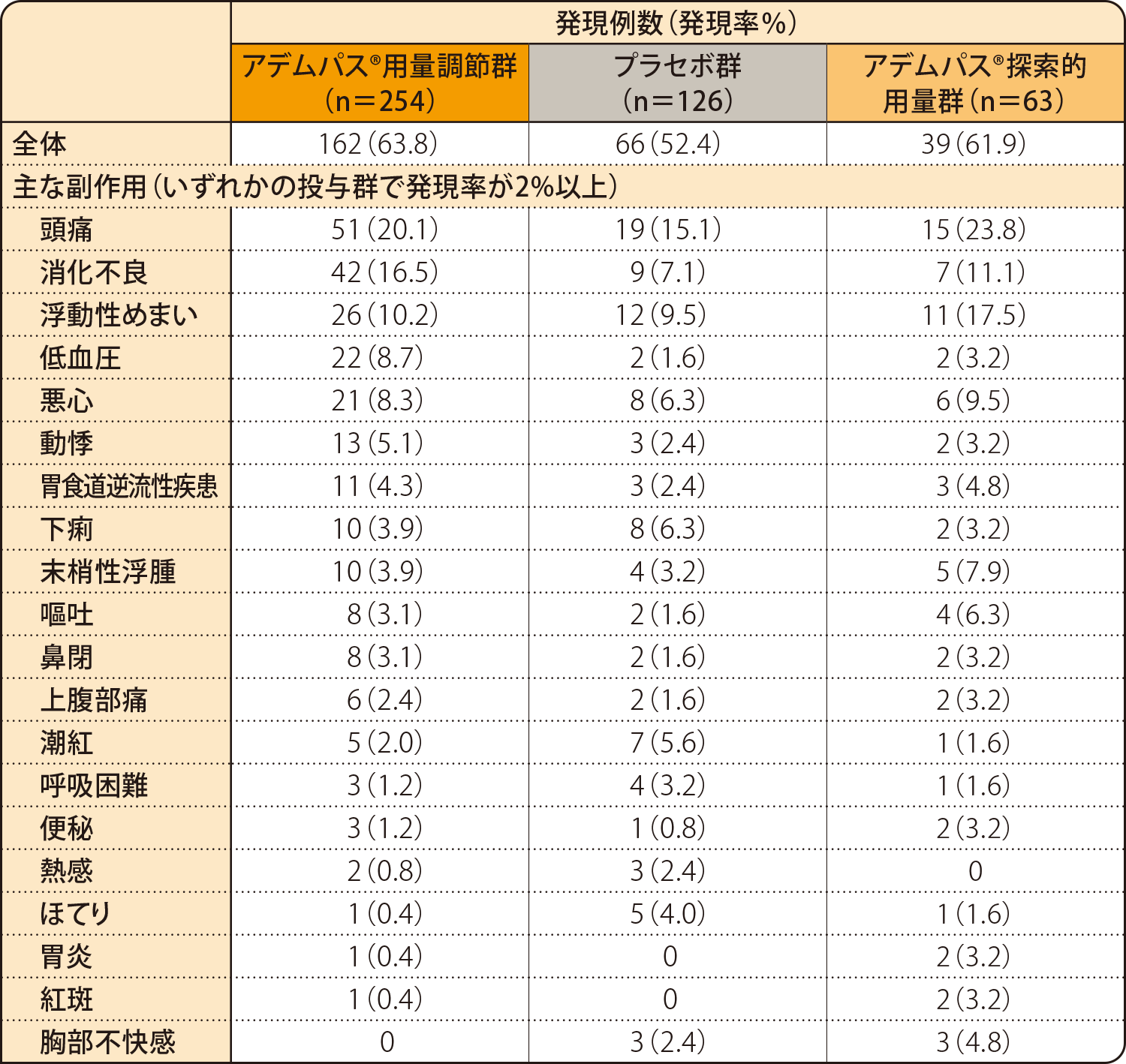
安全性解析対象集団
MedDRA version 15.0
副作用:治験薬と関連のある有害事象/治験薬投与下
重篤な副作用は、アデムパス®用量調節群で8例(3.1%)、プラセボ群で5例(4.0%)、アデムパス®探索的用量群で2例(3.2%)に認められました。その内訳は、失神がアデムパス®用量調節群で3例及びプラセボ群で1例、失神寸前の状態がアデムパス®用量調節群及びプラセボ群で各1例、肝酵素上昇、浮動性めまい、急性腎不全及び低血圧がアデムパス®用量調節群で各1例、下痢、呼吸困難及び肺動脈性肺高血圧症がプラセボ群で各1例、胃炎及び吐血がアデムパス®探索的用量群で各1例でした。
投与中止に至った副作用はアデムパス®用量調節群で6例(2.4%)、プラセボ群で5例(4.0%)に認められました。その内訳は、失神がアデムパス®用量調節群及びプラセボ群で各1例、上室性頻脈、食道浮腫、食道痛、全身性浮腫、肝酵素上昇、頚部痛、急性腎不全、低血圧がアデムパス®用量調節群で各1例、下痢、呼吸困難、低酸素症、肺動脈性肺高血圧症がプラセボ群で各1例でした。
本試験において死亡に至った副作用は認められませんでした。